
This program page is the compact operational companion to the research material on TranshumanGene. It summarizes the uploaded presentation into a web-readable mission brief.
Mission
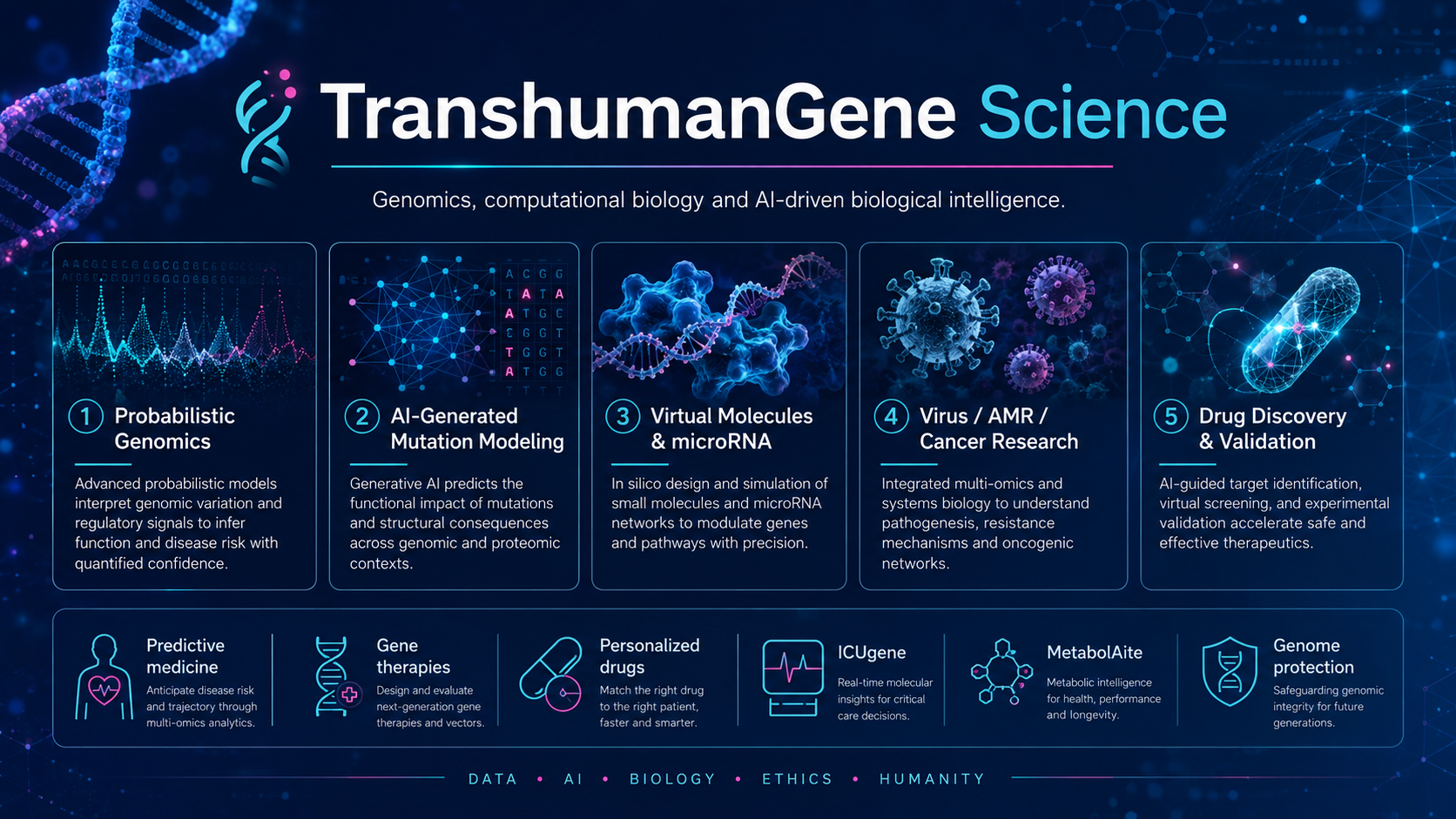
TranshumanGene explores how AI, supercomputing and computational biology can accelerate the organization of genomic data, the simulation of relevant mutations and the ranking of candidate molecules, microRNA and gene-therapy pathways.
Operational layers
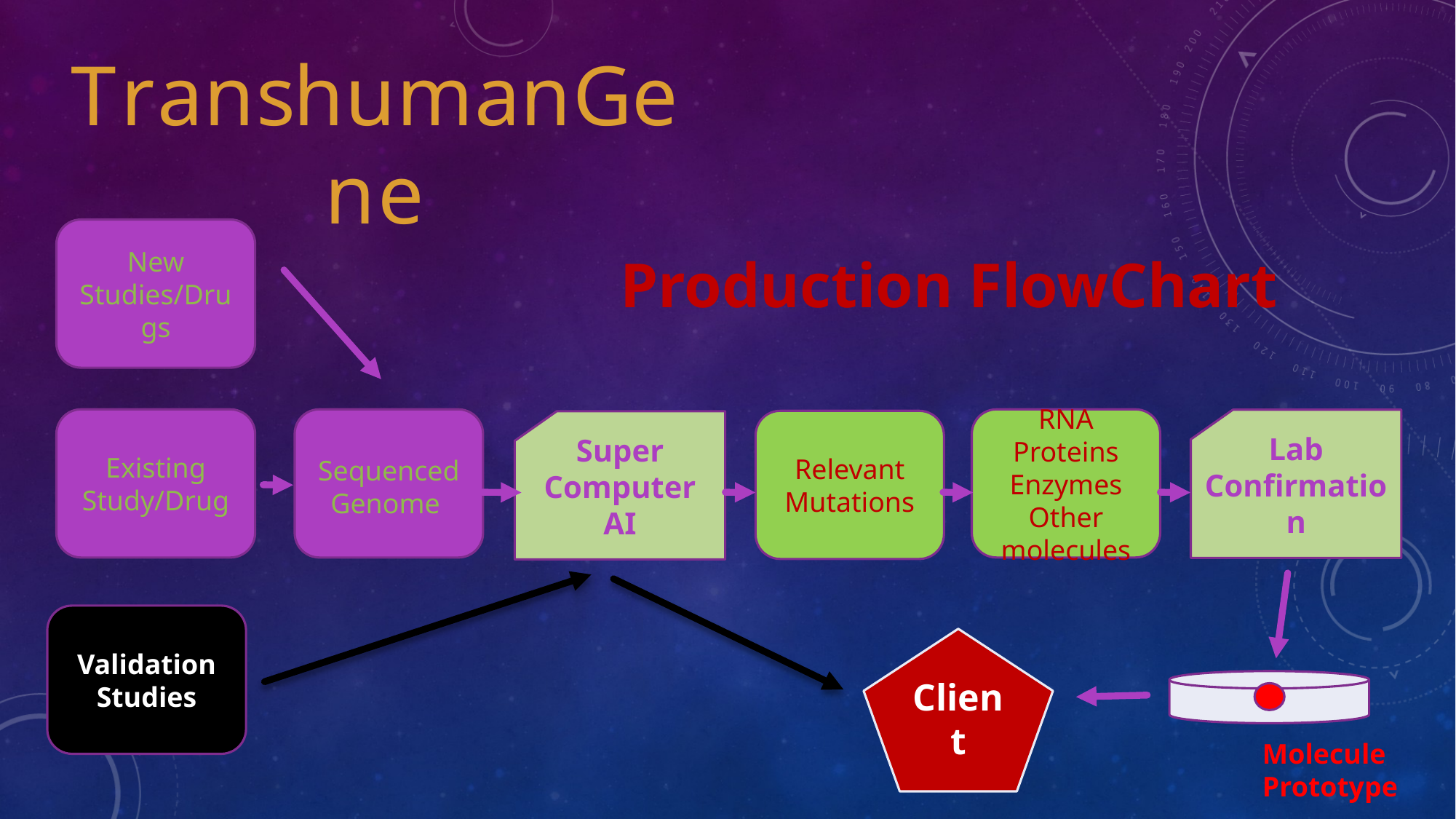
The program operates across four layers: data normalization, mutation modelling, candidate generation, and validation support. The work is meant to help teams decide what should be tested first, what should be deprioritized and how evidence can be documented more coherently.
Research tracks
Priority tracks include viruses, antimicrobial resistance, cancer-related studies, protein-level modelling, and translational biomedical education through OPENAIMED. The program also stresses global genomic inclusivity and the need for better calibrated datasets for underrepresented populations.
Related research pages
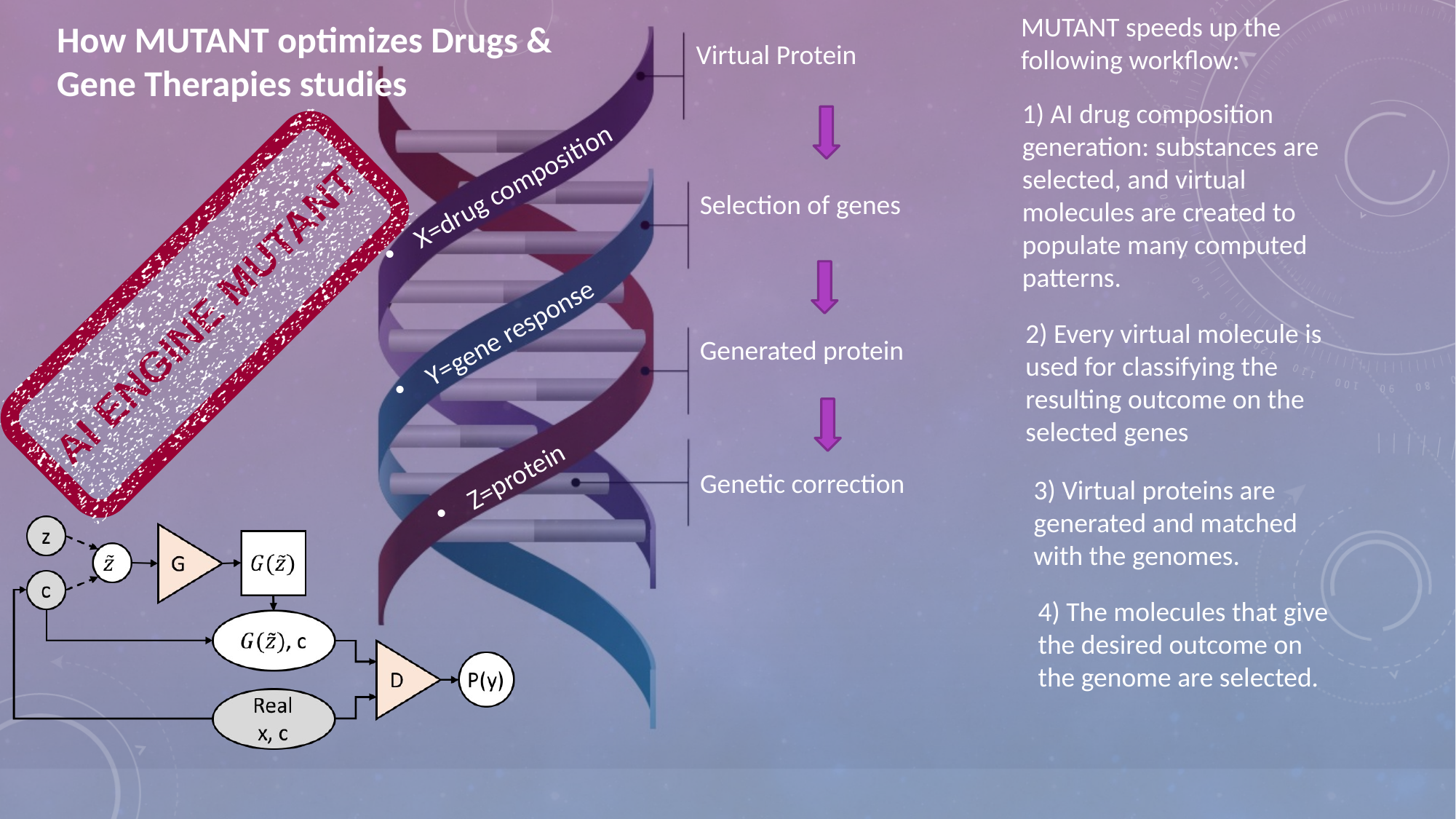
The three linked research modules below provide expanded explanations of probabilistic genomics, the MUTANT engine and the bio-digital engineering syllabus.